
Dr Kurt De Vos
PhD
School of Medicine and Population Health
Professor of Molecular and Cellular Neuroscience
+44 114 222 2241
Full contact details
School of Medicine and Population Health
Sheffield Institute for Translational Neuroscience (SITraN)
385a Glossop Road
Sheffield
S10 2HQ
- Profile
-
Dr De Vos studied chemistry and biotechnology and received a PhD in Biotechnology with greatest distinction from Ghent University, Belgium (1999; advisor Prof Johan Grooten). There he showed that clustering of mitochondria in the perinuclear region is an early event in apoptosis that is caused by inhibition of the molecular motor kinesin through hyperphosphorylation of the kinesin light chain (De Vos et al., 1998; De Vos et al., 2000).
He then embarked on his postdoctoral research work in the laboratories of Prof Mike Sheetz at Columbia University, New York, and Dr Vicky Allan at the University of Manchester. There he showed that phosphatidyl inositol phosphates control the direction of axonal mitochondrial transport (De Vos et al., 2003).
In addition he established that mitochondrial function controls mitochondrial dynamics and showed that the actin cytoskeleton is required for the recruitment of mitochondrial fission factor DRP1 to mitochondria (De Vos et al., 2005).
He became particularly interested in mitochondrial dynamics and neurodegeneration and relocated to the University of Sheffield to work in Dr Andy Grierson’s laboratory. There his research was focused on motor neuron diseases and the characterisation of mitochondrial axonal transport defects in models of amyotrophic lateral sclerosis (ALS) and hereditary spastic paraplegia (De Vos et al., 2007; Kasher et al., 2009).
This work was continued in the laboratory of Prof Chris Miller and Prof Chris Shaw in the MRC Centre for Neurodegeneration Research at King’s College London, and resulted in publications showing that VAPB interacts with mitochondrial protein PTPIP51 and that ALS VAPBP56S the disrupts axonal transport of mitochondria by increasing intracellular calcium levels.
End of 2011 he returned to Sheffield as faculty member in the newly established Sheffield Institute for Translational Neuroscience (SITRaN).
- Qualifications
-
- 2019 Reader in Molecular and Cellular Neuroscience
- 2016-2019 Senior Lecturer in Translational Neuroscience
- 2011-2015 Lecturer in Translational Neuroscience
- 2006-2011 Senior Researcher, MRC Centre for Neurodegeneration Research, The Institute of Psychiatry, King's College London, London, UK.
- 2004-2006 Postdoctoral Researcher, Academic Unit of Neurology, The University of Sheffield, Sheffield, UK.
- 2003-2004 Postdoctoral Researcher/Visiting Scientist, School of Biological Sciences, University of Manchester, Manchester, UK.
- 2000-2003 Postdoctoral Researcher, Department of Biological Sciences, Columbia University, New York, USA.
- 1994-1999 PhD in Science: Biotechnology (Greatest distinction), University of Ghent, Ghent, Belgium.
- Research interests
-
Research in the laboratory focuses on the mechanisms of nerve cell death in a number of neurodegenerative conditions, including motor neuron disorders such as amyotrophic lateral sclerosis (ALS; also known as motor neuron disease (MND) or Lou Gehrig disease) and hereditary spastic paraplegia (HSP), frontotemporal dementia (FTD), and Parkinson’s disease (PD). We are especially interested in the involvement of axonal transport, mitochondria and ER, and protein homeostasis/autophagy.
Current research themes include:
- The mechanisms causing defective axonal transport of mitochondria in ALS, PD and HSP.
- The cellular roles of the C9orf72 protein and their role in ALS and FTD
- The biology of close contacts between the endoplasmic reticulum (ER) and mitochondria and their involvement in health and disease
Work in the lab is funded by grants from the Medical Research Council (MRC), the Alzheimer’s Society, the Motor Neurone Disease Association (MNDA), the Thierry Latran Foundation, the Spastic Paraplegia Foundation, and the Moody Endowment Fund.
- Publications
-
Journal articles
- RuvBL1/2 reduce toxic dipeptide repeat protein burden in multiple models of C9orf72-ALS/FTD. Life Science Alliance, 8(2), e202402757-e202402757.


- A cell-penetrant peptide blocking
C9ORF72
-repeat RNA nuclear export reduces the neurotoxic effects of dipeptide repeat proteins. Science Translational Medicine, 15(685).
- Loss of TMEM106B exacerbates C9ALS/FTD DPR pathology by disrupting autophagosome maturation. Frontiers in Cellular Neuroscience, 16.
- C9ORF72-derived poly-GA DPRs undergo endocytic uptake in iAstrocytes and spread to motor neurons.. Life Sci Alliance, 5(9).
- Astrocyte adenosine deaminase loss increases motor neuron toxicity in amyotrophic lateral sclerosis. Brain, 142(3), 586-605. View this article in WRRO
- C9orf72 plays a central role in Rab GTPase-dependent regulation of autophagy.. Small GTPases, 9(5), 399-408. View this article in WRRO
- Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Human Molecular Genetics, 26(23), 4668-4679. View this article in WRRO
- C9orf72 Expansion Disrupts ATM-mediated Chromosomal Break Repair. Nature Neuroscience, 20, 1225-1235. View this article in WRRO
- SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nature Communications, 8. View this article in WRRO
- The Role of Mitochondria in Amyotrophic Lateral Sclerosis. Neuroscience Letters. View this article in WRRO
- Protein Homeostasis in Amyotrophic Lateral Sclerosis: Therapeutic Opportunities?. Front. Mol. Neurosci., 10, 123-123. View this article in WRRO
- C9ORF72 hexanucleotide repeat exerts toxicity in a stable, inducible motor neuronal cell model, which is rescued by partial depletion of Pten. Human Molecular Genetics, 26(6), 1133-1145. View this article in WRRO
- Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research?. Neurobiology of Disease. View this article in WRRO
- ALS/FTD-associated FUS activates GSK-3β to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations.. EMBO Rep, 17(9), 1326-1342. View this article in WRRO
- The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy.. EMBO J, 35(15), 1656-1676. View this article in WRRO
- Reduced number of axonal mitochondria and tau hypophosphorylation in mouse P301L tau knockin neurons.. Neurobiol Dis, 85, 1-10. View this article in WRRO
- Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations.. Nat Commun, 5, 5245. View this article in WRRO
- ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43.. Nat Commun, 5, 3996. View this article in WRRO
- Axonal Transport Defects in a Mitofusin 2 Loss of Function Model of Charcot-Marie-Tooth Disease in Zebrafish.. PLoS One, 8(6), e67276. View this article in WRRO
- Amyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria.. Hum Mol Genet, 21(9), 1979-1988. View this article in WRRO
- VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis.. Hum Mol Genet, 21(6), 1299-1311. View this article in WRRO
- Lemur tyrosine kinase-2 signalling regulates kinesin-1 light chain-2 phosphorylation and binding of Smad2 cargo.. Oncogene, 31(22), 2773-2782. View this article in WRRO
- A comparison of in vitro properties of resting SOD1 transgenic microglia reveals evidence of reduced neuroprotective function. BMC NEUROSCI, 12. View this article in WRRO
- A comparison of in vitro properties of resting SOD1 transgenic microglia reveals evidence of reduced neuroprotective function., 12(1), 91.
- Lemur tyrosine kinase-2 signalling regulates kinesin-1 light chain-2 phosphorylation and binding of Smad2 cargo, -.
- Phosphorylation of kinesin light chain 1 at serine 460 modulates binding and trafficking of calsyntenin-1., 124(Pt 7), 1032-1042.
- Amyotrophic lateral sclerosis mutant vesicle-associated membrane protein-associated protein-B transgenic mice develop TAR-DNA-binding protein-43 pathology.. Neuroscience, 167(3), 774-785.
- Amyotrophic lateral sclerosis mutant VAPB transgenic mice develop TDP-43 pathology.
- Deficiency of the copper chaperone for superoxide dismutase increases amyloid-β production. Journal of Alzheimer's Disease, 21(4), 1101-1105.
- Neurofilament subunit (NFL) head domain phosphorylation regulates axonal transport of neurofilaments., 88(4), 193-202.
- Riluzole protects against glutamate-induced slowing of neurofilament axonal transport., 454(2), 161-164.
- Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6.. Science, 323(5918), 1208-1211.
- Direct evidence for axonal transport defects in a novel mouse model of mutant spastin-induced hereditary spastic paraplegia (HSP) and human HSP patients.. J Neurochem, 110(1), 34-44.
- Role of axonal transport in neurodegenerative diseases.. Annu Rev Neurosci, 31, 151-173.
- Cell-free assays for mitochondria-cytoskeleton interactions.. Methods in Cell Biology, 80, 683-706.
- Visualization and quantification of mitochondrial dynamics in living animal cells.. Methods in Cell Biology, 80, 627-682.
- Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content.. Hum Mol Genet, 16(22), 2720-2728.
- Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. FASEB J, 20(9), 1407-1417.
- Mitochondrial function and actin regulate dynamin-related protein 1-dependent mitochondrial fission.. Curr Biol, 15(7), 678-683.
- Expression of phosphatidylinositol (4,5) bisphosphate-specific pleckstrin homology domains alters direction but not the level of axonal transport of mitochondria., 14(9), 3636-3649.
- RPTP-alpha acts as a transducer of mechanical force on alphav/beta3-integrin-cytoskeleton linkages.. J Cell Biol, 161(1), 143-153. View this article in WRRO
- Rap1 is involved in cell stretching modulation of p38 but not ERK or JNK MAP kinase.. J Cell Sci, 114(Pt 6), 1221-1227.
- Tumor necrosis factor induces hyperphosphorylation of kinesin light chain and inhibits kinesin-mediated transport of mitochondria.. J Cell Biol, 149(6), 1207-1214. View this article in WRRO
- Redox regulation of TNF signaling.. Biofactors, 10(2-3), 145-156.
- Significance of host cell kinesin in the development of Chlamydia psittaci.. Infect Immun, 67(10), 5441-5446.
- The 55-kDa tumor necrosis factor receptor induces clustering of mitochondria through its membrane-proximal region.. J Biol Chem, 273(16), 9673-9680.
- A caspase-activated factor (CAF) induces mitochondrial membrane depolarization and cytochrome c release by a nonproteolytic mechanism.. J Exp Med, 188(11), 2193-2198. View this article in WRRO
- Atractyloside-induced release of cathepsin B, a protease with caspase-processing activity.. FEBS Lett, 438(3), 150-158.
- Induction of unresponsiveness to tumor necrosis factor (TNF) after autocrine TNF expression requires TNF membrane retention.. J Biol Chem, 273(6), 3271-3277.
- Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proceedings of the National Academy of Sciences of the United States of America, 92(18), 8115-8119.
- An interaction between synapsin and C9orf72 regulates excitatory synapses and is impaired in ALS/FTD. Acta Neuropathologica.
Chapters
- Evidence of mitochondrial dysfunction in ALS and methods for measuring in model systems, International Review of Neurobiology Elsevier
- Role of Reactive Oxygen Species in Tumor Necrosis Factor Toxicity, Antioxidant and Redox Regulation of Genes (pp. 245-264). Elsevier
- Role of reactive oxygen species in TNF toxicity. In Sen KC, Sies H & Bauerle PA (Ed.), Antioxidant and Redox Regulation of Genes (pp. 245-264). Academic Press
Conference proceedings papers
- Cystatin C: The Forgotten Proteinopathy in ALS/MND. NEUROPATHOLOGY AND APPLIED NEUROBIOLOGY, Vol. 51
- Inosine reverses motor neuron toxicity observed in amyotrophic lateral sclerosis patient astrocytes with an adenosine deaminase deficiency. Biochimica et Biophysica Acta (BBA) - Bioenergetics, Vol. 1859 (pp e23-e23)
- The ER protein VAPB interacts with the mitochondrial protein PTPIP51 to regulate ER-mitochondria associations and calcium homeostasis. MOLECULAR BIOLOGY OF THE CELL, Vol. 24
- Mutant VAPB transgenic mice linked to amyotrophic lateral sclerosis type-8 develop TDP-43 pathology. BRAIN PATHOLOGY, Vol. 20 (pp 33-33)
- A14 Fast axonal transport of mitochondria is altered in Huntington's disease. Journal of Neurology, Neurosurgery & Psychiatry, Vol. 81(Suppl 1) (pp A5.1-A5)
- Mutations in RNA processing protein FUS result in neurodegeneration with cytoplasmic inclusion and cause familial amyotrophic lateral sclerosis. ACTA NEUROPATHOLOGICA, Vol. 118(3) (pp 445-445)
- Defective axonal transport of mitochondria in ALS. JOURNAL OF NEUROCHEMISTRY, Vol. 110 (pp 29-29)
- Mitochondrial shape is tightly linked to mitochondrial function. MOLECULAR BIOLOGY OF THE CELL, Vol. 13 (pp 127A-127A)
- Phosphatidylinositol (4,5) bisphosphate modulates mitochondrial motility in neurites. MOLECULAR BIOLOGY OF THE CELL, Vol. 12 (pp 166A-166A)
Software / Code
- CellCounter. http://rsbweb.nih.gov/ij/plugins/cell-counter.html: Retrieved from http://rsbweb.nih.gov/ij/plugins/cell-counter.html
Preprints
- C9orf72-derived poly-GA DPRs undergo endocytic uptake in iNPC-derived astrocytes and spread to motor neurons, Cold Spring Harbor Laboratory.
- A cell-penetrant peptide blocking C9ORF72-repeat RNA nuclear export suppresses neurodegeneration, Cold Spring Harbor Laboratory.
- Mitochondrial fission is increased in macrophages during mROS production in response to S. pneumoniae, Cold Spring Harbor Laboratory.
- LRRK2-mediated phosphorylation of HDAC6 regulates HDAC6-cytoplasmic dynein interaction and aggresome formation, Cold Spring Harbor Laboratory.
- RuvBL1/2 reduce toxic dipeptide repeat protein burden in multiple models of C9orf72-ALS/FTD. Life Science Alliance, 8(2), e202402757-e202402757.
- Research group
-
Current:
- Claudia Bauer (Postdoctoral Research Associate)
- Emma Smith (Postdoctoral Research Associate)
- Becky Cohen (PhD student)
- Lily Koryang (PhD student)
Previous:
- Anne-Kathrin Möller (Postdoctoral Research Associate)
- Kavitha Chinnaiya (Postdoctoral Research Associate)
- Chris Webster (PhD Student)
- Emma Wilson (PhD student)
- Richard Lucas (PhD student)
- Natalie Rounding (PhD Student)
- Yolanda Gibson (PhD Student)
- Gary Shaw (Research Assistant)
- Khlood Mehdar (PhD Student)
- Grants
-
Current:
Previous:
- Thierry Latran foundation
- Spastic paraplegia foundation
- Moody endowment fund
- Current projects
Axonal transport
Neurons are specialised cells in the brain and spinal cord that transmit nerve impulses (e.g. impulses of sensation to the brain, or impulses from the brain to muscles and organs). Neurons comprise cell bodies and long threadlike processes that extend away from the cell body. These processes are called axons and dendrites. Axons conduct impulses from the neuron cell body to other cells. In the brain, the end of the axon, called the synapse, connects to other neurons so as to mediate the brain’s computing power. In the spinal cord, motor neuron axons connect to muscle cells to facilitate muscle contraction. Axons of motor neurons can be longer than 1 meter!
Most of the proteins present in axons are produced in the cell body and are carried into and through the axon by a process called axonal transport. Molecular motor proteins drive axonal transport. These motors burn a fuel called ATP to generate locomotive force and run on protein tracks called microtubules. Thus, axonal transport is rather like a train journey with “locomotives” (molecular motors) that need “fuel” (ATP), run on “rails” (microtubules) and hook up to various “carriages” (cargoes). The components of axonal transport are shown in Figure 1.
Axonal transport of proteins and organelles is essential for proper neuron function and viability. When axonal transport malfunctions the axon slowly starves because the necessary proteins and other nutrients (e.g. ATP) are no longer delivered. The regions of the axon that are the furthest away from the cell body (motor neuron axons can be over a metre in length!) are most severely affected by axonal transport failure; these far-off axonal regions degenerate and die off, and as a result the connection between neurons and their targets is lost. Importantly, this is in fact what is observed in neurodegenerative diseases such as ALS.
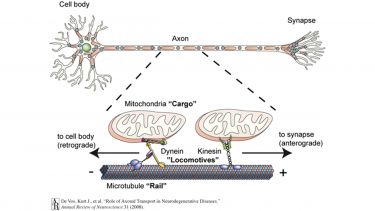
Figure 1: Axonal transport. Axonal transport of cargoes such as mitochondria is mediated by molecular motors (“Locomotives”) that run on microtubules (“Rails”). Kinesin molecular motors move toward the plus-end of microtubules and mediate transport toward the synapse. Cytoplasmic dynein moves toward the minus-end of the microtubule and mediates transport back to the cell body.
We now know that axonal transport malfunctions in a number of neurodegenerative diseases including ALS, HSP and PD (De Vos et al., 2007; Kasher et al., 2009). We also know that axonal transport malfunction is one of the earliest, and possibly the earliest defect in these diseases. Therefore, understanding how healthy axonal transport works and what causes it to malfunction in these diseases is very important and is likely to reveal novel drug targets that may be developed into medicines aimed at sufferers from these diseases.
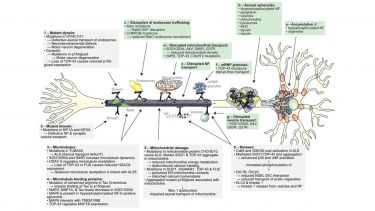
Figure 2. Axonal transport defects in ALS and underlying mechanisms. The axonal transport of various organelles has been shown to be defective in a number of ALS models and in ALS patients (a-g). A number of proposed molecular mechanisms underlying defective transport are indicated (1-6). See text for details. (Figure adapted with permission from Annual Review of Neuroscience, Vol. 31, De Vos, K. J., Grierson, A. J., Ackerley, S., and Miller, C. C., Role of axonal transport in neurodegenerative diseases, p151-173, Copyright © 2008 by Annual Reviews.) De Vos, K. J. & Hafezparast, M. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research. Neurobiol. Dis. 105, 283-299 (2017). DOI:10.1016/j.nbd.2017.02.004; CC BY 3.0.
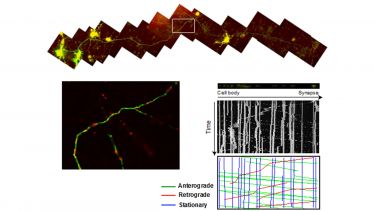
Figure 3: Measuring axonal transport. Neurons grown in culture on a coverslip (to panel) are transfected with green fluorescent protein (GFP) to visualize the axon and red fluorescent protein targeted to mitochondria to visualize mitochondria. Thus transfected neurons are put on a microscope and the movement of mitochondria is recorded. Movies of mitochondrial movements are converted into a kymograph to visualize movement.
Current projects in the lab investigate the molecular mechanisms of axonal transport defects in ALS, HSP and PD, and test novel drugs aimed at restoring axonal transport.
Recent findings from that lab are that:
- Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by PD-associated LRRK2 Roc-COR domain mutations (Godena et al, 2014).
- ALS-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels via actication of PINK1 (Moller et al., 2017)
The role of C9orf72, TMEM106B, autophagy and lysosomes in ALS/FTD
C9ALS/FTD –
A GGGGCC repeat expansion in intron 1 of the C9orf72 gene is the most common genetic cause of FTD and ALS (together referred to as C9FTD/ALS). The exact mechanisms by which the repeat expansion causes C9FTD/ALS are unknown, but there is evidence for both loss- and gain-of-function. Firstly, several observations suggest that C9orf72 loss-of-function by haploinsufficiency may be involved in C9FTD/ALS: (i) Reduced levels of C9orf72 mRNA have been reported in post-mortem tissue, patient-derived lymphoblastoid cell lines, iPSc and blood samples. (ii) Lower C9orf72 protein levels were detected in the frontal cortex of FTD and ALS cases. (iii) The GGGGCC repeat size correlates with transcriptional downregulation of the C9ORF72 promoter and onset age of disease. (iv) A loss-of-function splice site mutation in C9ORF72 has recently been identified in an ALS patient. (v) C9orf72 haploinsufficiency renders C9FTD/ALS MNs sensitive to glutamate excitotoxicity.Secondly, the pathogenic mechanism may involve a toxic gain-of-function via toxic repeat RNA species and/or repeat-associated non-ATG (RAN) translation of the repeats into toxic aggregation-prone dipeptide-repeat proteins (DPRs) that inhibit nuclear import/export pathways.
Knockout of C9orf72 in mice does not cause overt neurodegeneration but mild motor deficits have been reported, suggesting that haploinsufficiency is involved in disease but is not sufficient to cause full C9FTD/ALS per se. Likewise, BAC transgenic mice harbouring a human GGGGCC C9orf72 repeat expansion show no or only mild signs of neurodegeneration despite proven presence of RNA foci and DPRs, suggesting that RNA and DPR toxicity are not sufficient to cause C9FTD/ALS either. These observations in mouse models together with the evidence for all three toxic pathways in patients and patient-derived cell models strongly indicate that both loss- and gain-of-function mechanisms contribute to pathogenesis in a multiple-hit fashion.
C9orf72 is a regulator of Rab GTPases in the autophagy pathway –
C9orf72 is structurally related to Differentially Expressed in Normal and Neoplasia (DENN) domain-containing proteins, which function as GDP/GTP exchange factors (GEFs) for the Rab family of GTPases. Rab GTPases function as molecular switches that alternate between a GTP-bound active state and a GDP-bound inactive state. GEFs catalyse the exchange of GDP for GTP while GTPase activating proteins (GAPs) catalyse GTP hydrolysis to GDP. GTP-bound, active Rabs recruit and/or activate a variety of effector molecules and in doing so Rabs control a range of cellular trafficking events, including autophagy.We have recently shown that the C9orf72 protein regulates autophagy initiation via Rab1a. C9orf72 mediates the interaction between Rab1a and the ULK1 autophagy initiation complex and facilitates trafficking of the ULK1 autophagy initiation complex to the phagophore. Consequently, depleting C9orf72 levels using siRNA significantly inhibited autophagy (Webster et al., 2016; Webster et al., 2018). Others have reported similar findings in vitro and in vivo. Importantly, induced neural progenitor cell (iNPC)-differentiated neurons (iNeurons) derived from C9FTD/ALS patients show deficits in basal autophagy levels, confirming possible relevance in disease (Webster et al., 2016).
C9orf72 forms a complex with SMCR8 and WDR41 and this complex acts as a GEF for Rab8a and Rab39b that are involved in the maturation stages of autophagy. The interaction between the C9orf72/SMCR8/WDR41 complex and Rab8a or Rab39b is mainly through binding with SMCR8, and Rab GEF activity was only observed when SMCR8 was present. As SMCR8 is itself a DENN protein it appears that SMCR8 rather than C9orf72 may mediate the GEF activity toward Rab8a and Rab39b.
Sequential activation of Rab GTPases in a cascade like fashion allows the correct spatial and temporal recruitment of successive Rabs in a signal transduction pathway. In these Rab cascades the upstream Rab and its effectors recruit the GEF of the next downstream Rab, while the downstream Rab recruits the GAP for the upstream Rab. Thus, C9orf72 links autophagy initiation to downstream events via a Rab cascade, involving Rab1a upstream of Rab8a and Rab39b (Fig. 1; Webster et al., 2018).
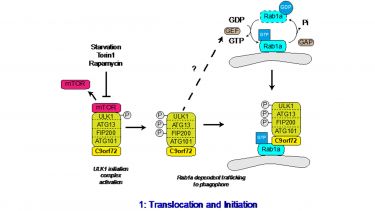
Figure 1. C9orf72 regulates the Rab1a dependent trafficking of the ULK1 complex. Upon inhibition of mTOR, the inhibitory phosphorylation of ULK1 is lost, leading to the activation of ULK1. Active ULK1 phosphorylates the other members of the initiation complex, including FIP200 and ATG13. Functioning as a Rab1a effector, C9orf72 mediates the interaction between the active ULK1 initiation complex and GTP-Rab1a within its target membrane. Thus C9orf72 controls the Rab1a dependent trafficking of the ULK1 initiation complex to the phagophore. How Rab1 is activated in response to autophagy induction is not yet known. © 2016 The Author(s). Published with license by Taylor & Francis© Christopher P. Webster, Emma F. Smith, Andrew J. Grierson, and Kurt J. De Vos; CC BY 3.0; https://doi.org/10.1080/21541248.2016.1240495
Proper function of autophagy and the endo-lysosomal systems are thought to be crucial for neuronal health, and dysfunction has been connected to neurodegeneration. Indeed, neuron-specific knockout of the essential autophagy genes ATG5 or ATG7 cause neurodegeneration in mice. In addition to C9orf72 a large number of other ALS/FTD associated genes are implicated in the autophagy pathway (Fig. 2; Webster et al., 2018), leading to the hypothesis that loss of autophagy may contribute to the development of the disease.
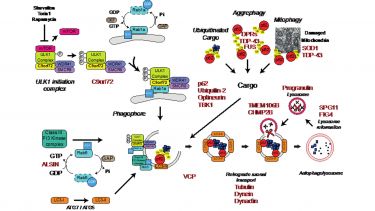
Figure 2. Autophagy and ALS/FTD. A number of genes, indicated in red, implicated in ALS/FTD have been linked to the autophagy/lysosomal pathway. © 2016 The Author(s). Published with license by Taylor & Francis© Christopher P. Webster, Emma F. Smith, Andrew J. Grierson, and Kurt J. De Vos; CC BY 3.0; https://doi.org/10.1080/21541248.2016.1240495
Current projects in the lab aim to further understand the cellular functions of the C9orf72 protein in autophagy, its possible role in ALS/FTD, and its relation with the ALS/FTD risk factor gene TMEM106B.
Mitochondria-associated ER membranes (MAM)
Mitochondria-associated ER membranes (MAM) are specialized domains of the endoplasmic reticulum (ER) that are in physical contact with mitochondria. Between 5 and 20% of the mitochondrial surface is closely apposed to MAM membranes. Through these close contacts, mitochondria and ER communicate directly with each other via the exchange of calcium signals. Under physiological conditions, mitochondrial calcium activates the rate-limiting enzymes of the Krebs cycle and thereby increases oxidative phosphorylation and ATP synthesis to match local energy demand. In turn, energized mitochondria influence ER calcium homeostasis and redox dependent ER processes such as oxidative protein folding. In agreement with an essential cellular function of ER-mitochondria communication, dysfunctional signalling between ER and mitochondria has been linked to neurodegenerative diseases, diabetes, cancer and inflammation.
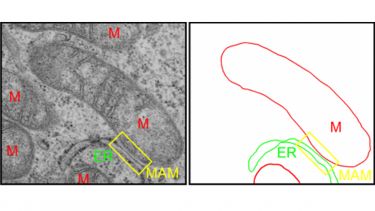
Figure 3: Visualization of MAM by transmission electron microscopy. Mitochondria are labelled with “M”.
A large body of evidence implicates ER stress and damage to mitochondria in the pathogenesis of both familial and sporadic ALS. However, the upstream causes of ER stress or mitochondrial damage remain to be determined. How mutations in non-ER and non-mitochondrial proteins that cause ALS elicit ER stress and mitochondrial dysfunction is a conundrum and the mechanisms linking these apparently disparate insults with other ALS-associated pathologies such as cytosolic TDP43 accumulation and defects in axonal transport are unclear.
Our data suggests that a possible explanation lies in dysfunctional signalling at MAM. We have shown in two publications that MAM is implicated in familial ALS caused by a mutation in VAPB (VAPBP56S). We found that perturbation of MAM by VAPBP56S causes mitochondrial dysfunction by calcium overload and as a consequence disturbs calcium homeostasis (De Vos et al., 2012). This in turn causes a permanent increase in cytosolic calcium levels, which is directly responsible for the inhibition of anterograde transport of mitochondria in VAPBP56S-expressing neurons (De Vos et al., 2012; Morotz et al., 2012)(see above axonal transport). Thus, our data shows that damage to inter-organelle signalling at the ER-mitochondria interface is upstream of a number of ALS-associated toxicity pathways including mitochondrial dysfunction, disturbance of calcium homeostasis, and defective axonal transport.
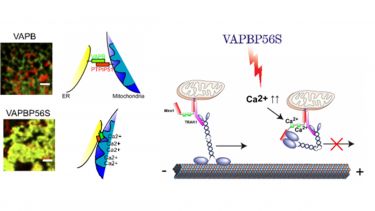
Figure 4: VAPBP56S disrupts axonal transport. VAPBP56S increases interaction of ER and mitochondria via PTPIP51. This leads to calcium overload in mitochondria and an increase in cytosolic calcium levels. Increased cytosolic calcium binds to Miro1 and inhibits kinesin-mediated anterograde transport of mitochondria.
Current projects further investigate these findings and extend these studies to other models of ALS to establish if compromised MAM and defective signalling between ER and mitochondria is a common phenomenon in ALS and a possible therapeutic target.






